The Coming Alzheimer’s Epidemic, the Cholinergic Hypothesis, and Nerve Growth Factor Gene Therapy
Alzheimer’s disease (AD) is the top cause of dementia, and the top risk factor for AD is age–with almost half of people above the age of 85 suffering from the disease.
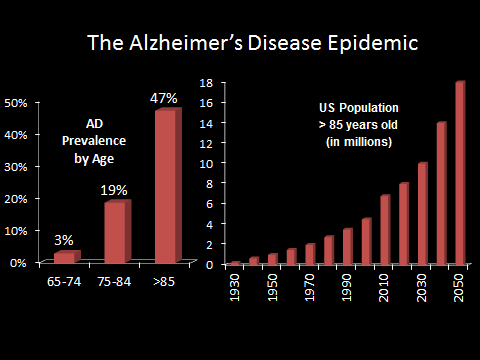
With the aging baby boomer generation and medical advances squaring off the life expectancy curve, the United States is projected to have over 9 million cases of AD by 2050. This huge expected increase in cases has been dubbed by some as the “Alzheimer’s Disease Epidemic.”
Alzheimer’s disease is marked primarily by memory loss and inability to form new memories, but it can also impair language, higher thinking, visuospatial skills, and it can even cause personality changes and delusions. By erasing patients’ identities, memories, and personalities, Alzheimer’s disease can rob patients of their humanity, and can be devastating for the friends and family watching the disease unfold.
 Auguste D.’s story
Auguste D.’s story
Alzheimer’s disease was first described in 1901, a patient Auguste D., a 51-year-old woman, suffering from debilitating memory loss, deficits, and persecutory delusions. She underwent a progressive decline and died five years later. The case was described by Alois Alzheimer a German psychiatrist and neuropathologist (though now the disease is treated by neurologists).
The fact that Alzheimer was a pathologist was of crucial importance. Because of it, he performed an autopsy on Auguste D. and discovered proteinaceous plaques, and neurofibrillary tangles in the woman’s brain, which to this day remain the pathological hallmarks of Alzheimer’s disease:
Alzheimer’s mentor, Kraepelin, was the first to use the eponym “Alzheimer’s Disease” in 1904, and his writing about the disease was prescient: “Although the anatomical findings suggest that we are dealing with a particularly serious form of senile dementia… this disease sometimes starts as early as in the late forties.”
This clinical observation was later borne out by genetic studies, while most people get non-familial AD which appears to have complex genetic and environmental risk factors in old age, there is an early-onset familial variant of AD that affects other-wise normal, healthy patients in their 40’s or even younger. This familial variant is caused by autosomal dominant rare mutations, so someone suffering from familial AD has a fifty percent chance of passing it on. Familial AD is devastating, but only accounts for only a tiny minority of cases.

Intriguingly, about the first three genes identified that cause familial Alzheimer’s disease were all part of the same biological pathway. These genes code for the amyloid precursor protein (APP), and presenilin 1 and 2. Amyloid precursor protein is cleaved by the “gamma secretase complex,” an enzyme that is made up of multiple subunits, including presinilin 1 & 2. If APP is cleaved at the wrong locations, it forms a protein fragment beta-amyloid, which can induce other proteins to misfold, forming a protein aggregate that grows like a snowball rolling down a hill. This protein aggregate eventually forms a giant extracellular plaque—the very same plaques that Alzheimer’s described in his patient Auguste D!
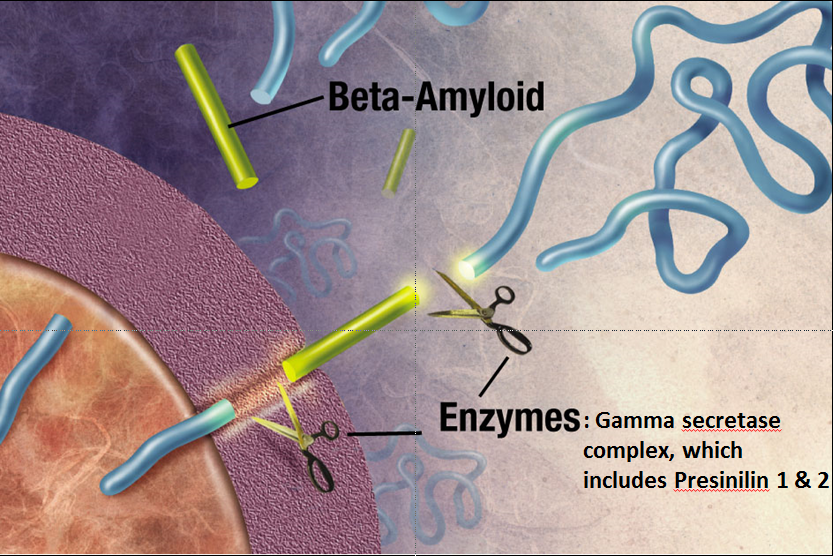

The Amyloid Cascade Hypothesis
This genetic work led to the amyloid cascade hypothesis: that extracellular amyloid accumulates and initiates a sequence of events, eventually leading to neurotoxicity and clinical symptoms in AD. This hypothesis has inspired a number of clinical trials to test drugs to treat or slow the course of Alzheimer’s, by attempting to stop the formation or remove these beta-amyloid plaques. Even when trials have been successful y removed patient’s plaques, patients have not shown clinical improvements. It is unclear whether the amyloid cascade hypothesis is wrong (and perhaps amyloid plaques correlate with but do not cause AD), or whether we are just starting our treatments too late once the cascade of neurodegeneration is initiated–but that these treatments could be successful if we used them on patients earlier in their disease course.
In fact we know that patients with Alzheimer’s disease don’t show clinical symptoms until late in the disease, perhaps due to a phenomenon known as cognitive reserve. New techniques however, allow us to determine whether people showing subtle cognitive deficits are at high or low risk of developing Alzhiemer’s, and therefore we can continue to test the amyloid cascade hypothesis by performing clinical trials using the drugs we know clear plaques on people who are of high risk of developing Alzheimer’s.
For example, the same proteins found in the plaques and tangles in Alzheimer’s disease brains such as amyloid beta make their way into the cerebrospinal fluid which coats the brain and spinal cord. In patients with mild cognitive impairment–meaning they are starting to show cognitive problems compared to age-matched, education-matched controls, but not severe enough to be classified as Alzheimer’s–we can perform lumbar punctures to harvest their cerebrospinal fluid. Then by measuring levels of these proteins found in plaques, we can predict which of these patients with MCI are at high risk of progressing to Alzheimer’s.
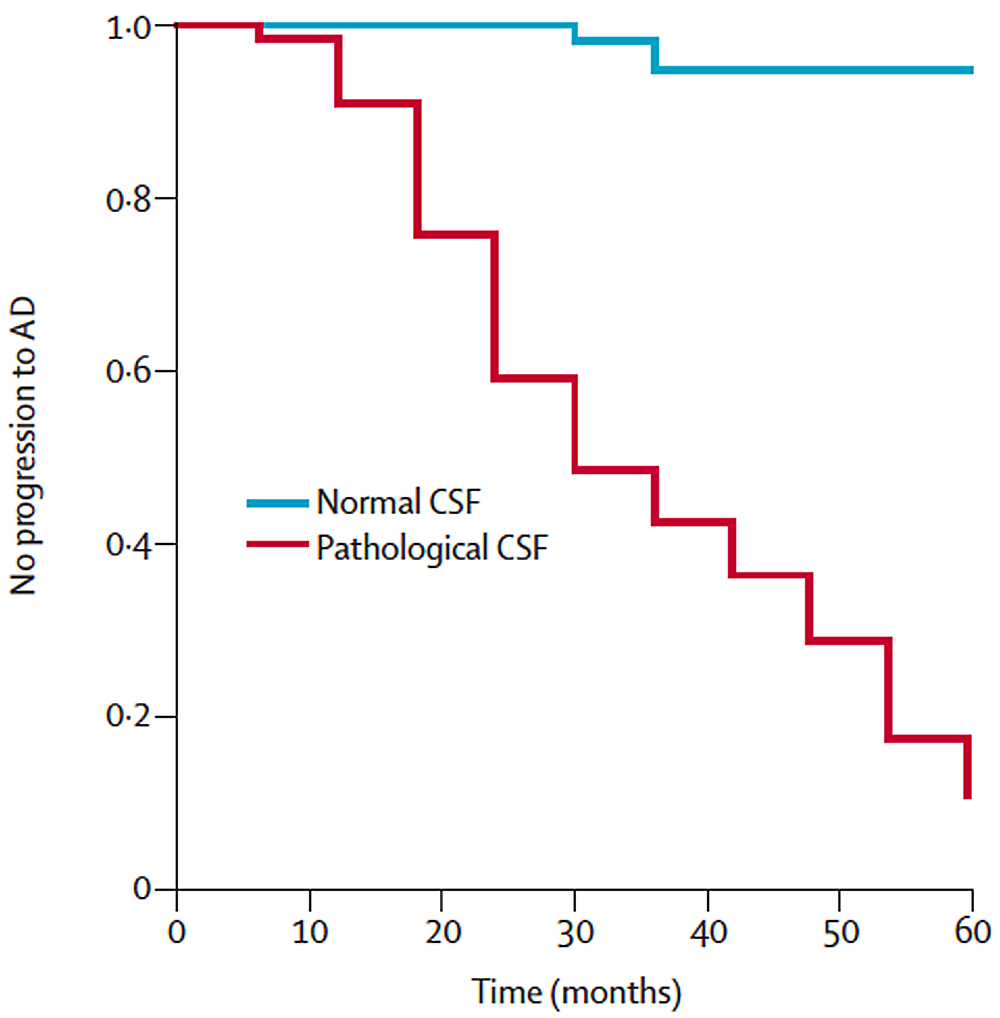 Hansson et al., Lancet Neurol (2006)
Hansson et al., Lancet Neurol (2006)
Additionally, we can look at the burden and distribution of plaques in the brain using radioactive PET ligands that bind to the plaques:

And we are developing techniques to non-invasively visualize and quantify the burden of this plaques in retina: 
The Cholinergic Hypothesis
While the previously-described techniques will inevitably assist with the diagnosing AD, the amyloid cascade hypothesis is still unproven, and so far has not lead to any effective therapy. Currently, one of the best treatments for Alzheimer’s disease is lifestyle modification: labeling and arranging one’s life as memory declines. The mainstay of pharmacological approach for AD are cholinesterase inhibitors developed in the 1990s and 2000s, which can improve performance on cognitive tests.

These drugs block acetylcholinesterase, blocking the breakdown of the neurotransmitter acetylcholine into acetate and choline, so the drugs therefore they increase levels of acetylcholine in the synapse and signaling to the postsynaptic cell.
You might be wondering, how do these drugs help treat Alzheimer’s if it is a disease caused by plaques, tangles and neurodegeneration? Why did we develop and test them in the first place?
These drugs were actually developed based on an earlier theory of Alzheimer’s the cholinergic hypothesis.
In the 1960s and ‘70s psychologists gave young healthy subjects cholinergic inhibitors (which prevent acetylcholine from signaling to post-synaptic cells). These young healthy subjects had memory problems and cognitive problems that resembled patients with Alzheimer’s disease, showing that acetylcholine was important for memory.

Most of the brain’s supply of acetylcholine derives from the nucleus basalis of Meynert in the basal forebrain, so doctors investigated this area during the autopsies of patients with Alzheimer’s and showed that the nucleus basalis seems to be particularly vulnerable to the neurodegeneration seen in AD. Patients showed decreased numbers of healthy neurons in this area, and decreased cholinergic projections to the hippocampus and entorhinal cortex (areas we also knew were important for memory), suggesting that patients with AD may have decreased acetylcholine.
Going back to the drugs we use for Alzheimer’s cholinesterase inhibitors, these drugs are thought to boost the remaining acetylcholine signaling going on in synapses, similar to how SSRIs increase the amount of serotonin. However, because they are not preventing the neurodegeneration that is causing AD, disease progression continues and causes massive neurodegeneration, eventually killing patients by interfering with their ability to swallow and breath.

However, if the cholinergic hypothesis is correct, and disruptions to the acetylcholine signaling from the basal forebrain is especially important for the symptoms of AD, then maybe if we can selectively prevent degeneration of this area we can slow the disease. Currently clinical trials are delivering of Nerve Growth Factor (NGF) to the basal forebrain using gene therapy approaches—implanting stem cells that produce NGF or using viruses to modify cells in the basal forebrain to produce it themselves. Nerve Growth factor is known to increase the growth and vitality of cholinergic neurons, and may therefore help preserve the cells as they suffer early insults of Alzheimer’s.
Even if Nerve Growth Factor gene therapy can just slow the disease, we can give patients more time with their memories intact to enjoy their families and a dignified independent life. We can decrease the time families and friends spend caring for a person who’s face the recognize, but whose mind becomes increasingly unfamiliar, and we can decrease the time patients have to spend institutionalized or with a full time caretaker.
Clinical research is slow, and research is never certain, but the new approaches to Alzheimer’s give hope to a Alzheimer’s disease—a disease where prognoses for patients like Auguste D. have remained gloomy for over a century, and where an epidemic of new cases looms ahead in the near future.
Filed under: Uncategorized | Leave a Comment
-

Subscribe via RSS
Current Neuroscience Book Recommendation:
In Search of Memory -Eric Kandel
Kandel’s book is a nobel prize winner's memoir that spans from his Jewish childhood in Nazi-occupied Vienna to his work on sea slugs that uncovered synaptic plasticity, the molecular foundation of learning and memory. When I graduated college, every neuroscience major was given a copy of this book.


No Responses Yet to “The Coming Alzheimer’s Epidemic, the Cholinergic Hypothesis, and Nerve Growth Factor Gene Therapy”